
國際罕見病日 | 當罕見病遇見新生兒篩查,一切有了答案
發(fā)布時間:2021/2/28 10:56:09??/??瀏覽量:??/??【關閉】
每年二月的最后一天為國際罕見病日,今年是第14個國際罕見病日,今年的主題是“Rare is many. Rare is strong. Rare is proud.”——“罕多,罕強,罕驕傲”。

罕見病日主題故事:6大洲,6張面孔,6位英雄,6條生命。來自全球不同地方不同年齡的6位罕見病患者講述了他們的故事,Angelina來自澳大利亞,患有X連鎖腦橋小腦發(fā)育不全伴智力低下和小頭畸形(MICPCH)。Tristan 是美國的時裝設計師,患有鐮狀型細胞貧血癥。Regina來自巴西,患有平滑肌肉瘤。Syafiq 來自馬來亞,患有少汗型外胚層發(fā)育不良癥(HED)。Harvey來自肯尼亞,患有脊髓性肌萎縮。Jon-Kristian生于挪威,患有成骨不全癥。
(一)罕見病并不罕見
罕見病,是一大類發(fā)病率很低、大多數具有遺傳性的疾病,又稱“孤兒病”。事實上罕見病并不罕見,世界衛(wèi)生組織數據顯示,罕見病占人類疾病的10%,約有4億人飽受罕見病的困擾。中國罕見病患者總數約為1680萬,在人群中的綜合發(fā)病率超過1%。目前已經明確的罕見病有7000多種,其中80%為遺傳病。
√新生兒篩查科 是集新生兒遺傳代謝病篩查與隨訪、診治于一體的專業(yè)科室,具備雄厚的技術力量、優(yōu)良的人才梯隊、完善的隨訪系統(tǒng)和先進的設施、設備。
新生兒篩查科成立于1996年,是青島市衛(wèi)健委批準的青島市唯一一家新生兒先天性遺傳代謝性疾病檢測與診治機構,承擔全市新生兒篩查、隨訪與診治工作。中心擁有先進的串聯質譜檢測系統(tǒng)、全自動時間分辨熒光檢測儀。中心成立伊始即開展先天性甲狀腺功能低下(CH)、苯丙酮尿癥(PKU)、腎上腺皮質增生癥(CAH)和葡萄糖-6-磷酸脫氫酶(G6PD)缺乏四種常規(guī)疾病的新生兒篩查。自2011年底始開展串聯質譜技術(MSMS)篩查新生兒遺傳代謝病,檢測范圍涉及氨基酸代謝障礙、脂肪酸氧化代謝障礙、有機酸代謝障礙等40余種遺傳代謝病的篩查工作,通過篩查,使得部分先天性遺傳代謝病可治、可防、可干預,提高其生活質量,同時避免先證家庭同樣患兒的出生。新生兒篩查中心將在現有的基礎上,繼續(xù)推進全市遺傳代謝病的篩查工作,同時加強與臨床科室的聯系,對臨床上無法明確原因的患兒進行輔助檢查,明確其病因,得到早期的診治。
(二)罕見病與新生兒篩查的“親密關系”
2018年6月8日,國家衛(wèi)生健康委員會等五部委聯合制定的《第一批罕見病目錄》正式發(fā)布,涵蓋了共121種罕見病,其中有40多種已經有批準上市的治療藥物。我們注意到,這121種罕見病被納入的條件,與WHO新生兒篩查病種的準入國家標準(Wilson and Junger原則)居然不謀而合:
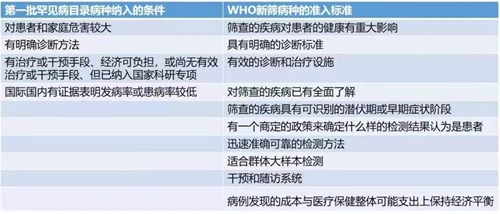
我國最早納入新生兒篩查的病種是先天性甲狀腺功能減低癥和苯丙酮尿癥,生化和質譜技術的發(fā)展又推動先天性腎上腺皮質增生癥、葡萄糖-6-磷酸脫氫酶缺乏癥、氨基酸、有機酸和脂肪酸類代謝病逐漸納入新生兒篩查疾病譜。隨著分子技術和二代測序的發(fā)展,以及治療遺傳病的新藥不斷涌現,還可以進行糖原累積癥、肝豆狀核變性、重癥聯合免疫缺陷病、血友病、耳聾、溶酶體病、杜氏肌營養(yǎng)不良癥等疾病的篩查。隨著篩查病種的不斷增加,更多的罕見病可以在早期發(fā)現,并在出現嚴重癥狀前給予早期干預,能夠很大程度上縮短疾病診斷時間,降低診斷成本,減輕疾病對患者及其家庭身體、精神以及經濟上的傷害。

(三)溶酶體貯積癥 因治療藥物納入醫(yī)保而廣受關注
溶酶體貯積癥(LSDs),是一組由先天代謝缺陷引起的罕見疾病,疾病種類多達50多種,包括法布里病、戈謝病、糖原貯積病Ⅱ型(又名龐貝氏病)、尼曼-匹克病等,聯合發(fā)病率約為1/8,000。LSDs會引起周圍神經病變,發(fā)育遲緩,進行性智力、運動倒退,骨骼異常等,甚至致命,對患者和家庭造成極大的經濟和精神負擔。然而大部分患者都曾有過誤診的經歷,并且往往需要輾轉多家醫(yī)院花費數年時間才能確診,因而貽誤了最佳治療時機。而通過新生兒篩查實現早期篩查、早期診斷、早期治療是避免誤診,改善預后,挽救這些患兒的關鍵。
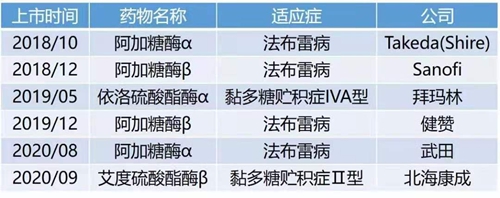
近年來,越來越多的溶酶體貯積癥的治療方案及治療藥物獲得突飛猛進的發(fā)展。目前已有的LSD治療方法包括酶替代療法、造血干細胞移植、底物減少療法等,其中已獲美國FDA或歐洲EMA批準上市的部分LSD治療藥物有近20種;過去的三年,我國也分別審批上市了兩種藥品用于治療法布雷病、黏多糖貯積癥II型、黏多糖貯積癥IVA型,為國內LSD患者帶來福音。
由于這些罕見病藥物需要長期服用且藥價極高,包括全國人大代表在內的社會各方力量竭力奔走呼吁:在全國范圍內將戈謝病納入醫(yī)保目錄!在大家的努力下,LSDs患者終于迎來了曙光:部分自身條件較好的省份首先將戈謝病、龐貝病納入醫(yī)保;藥企、科研人員加快戈謝病等新藥、新療法的研發(fā);國家也一直在推動通過大病保險、醫(yī)療救助、財政專項等方式,解決戈謝病的用藥困局。
溶酶體貯積癥納入新篩病種,不再遙遠
隨著研究的不斷深入,干預和治療方案日趨成熟和規(guī)范,美國、意大利的部分地區(qū)已經將部分病種納入常規(guī)的新生兒篩查項目,巴西、法國、德國、南非等國家也已經開展試點研究。目前,國內山東等省份也已經開始了溶酶體貯積癥新生兒篩查的試點研究。2020年6月20日,由北京健康促進會主辦,珀金埃爾默和賽諾菲協辦的溶酶體貯積癥高危篩查項目正式啟動。2020年11月21日中國溶酶體貯積癥新生兒篩查協作組成立并開展了2020年國際溶酶體貯積癥高峰論壇。社會各方及學術組織的努力,讓溶酶體貯積癥納入新生兒篩查已不再遙遠。
DMD:鐘情于男性的肌營養(yǎng)不良癥
DMD——杜氏肌營養(yǎng)不良癥,又稱假肥大型進行性肌營養(yǎng)不良,為X連鎖隱形遺傳病,是肌營養(yǎng)不良癥中最嚴重最常見的類型,發(fā)病率為1/3000-1/6000活產男嬰。DMD患者最初的癥狀通常出現在3-5歲,包括運動遲緩、步態(tài)異常、站立困難及易跌倒等,少數患者還可表現出語言發(fā)育遲緩或全面性發(fā)育落后。隨時間逐漸加重喪失獨立活動能力,十幾歲時需坐輪椅,并可能出現危及生命的心臟和呼吸系統(tǒng)疾病,通常在20-30歲時死亡。DMD患者的確診時間一般需要耗費近5年的時間,通過早期的DMD篩查可以縮短確診時間,改善患者預后,指導患者父母的再生育。
基因治療,拯救DMD患者的希望
對于DMD的治療,目前尚未有治愈的手段。臨床主要通過藥物治療、物理治療、基因治療、以及干細胞治療的手段對疾病進行控制。近年來,相繼有多款新藥獲批上市,為患者提供了更多的臨床用藥選擇。同時,多種治療手段均在臨床試驗各階段當中,為疾病的治療和治愈點燃了希望。美國FDA 2020年已獲批的53款藥品中,13款創(chuàng)新藥主要用于治療罕見病或孤兒病,其中就包括適用于53號外顯子跳躍杜氏肌營養(yǎng)不良患者的Viltepso(viltolarsen)。
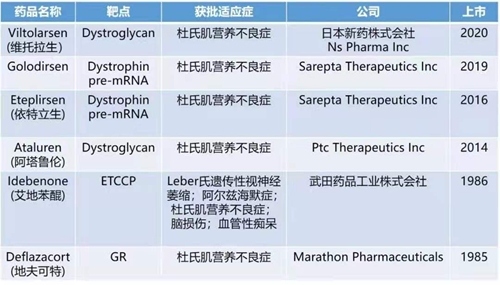
近年全球獲批上市的DMD治療藥物
更早推動DMD新生兒篩查的必要性
Annemieke等人的15項有關診斷時間減少和懷疑DMD后續(xù)診斷方法的調研內容顯示,11項議題中接近100%的家庭強烈認同并推崇,4項內容得到80%以上家庭的認可和推崇,由此可見,縮短DMD確診時間,推動更早篩查已勢在必行。
1975-2011年間,全球多個國家和地區(qū),通過檢測干血斑樣本中肌酸激酶的水平,完成了180多萬新生兒的篩查,發(fā)病率為1/4500。美國疾病控制和預防中心也建議,早期診斷可以使DMD患者得到更個性化的治療。因此,DMD的新生兒篩查也是大勢所趨,通過新生兒早期篩查,可以避免誤診,縮短確診時間,延緩患兒病情發(fā)展,有更多的時間等待治療藥物,也可以更好的指導DMD患者父母再生育。
隨著政府及社會力量對罕見病防治工作的支持力度不斷加大,更多的罕見病能夠通過新生兒篩查公共衛(wèi)生體系達到早篩查、早診斷、早治療的效果,降低這些疾病對整個人類群體的危害,提高全人類的健康水平和生活質量。